
Penn Vet News
Featured News

Penn Vet’s Wildlife Futures Seek to Unravel the Mystery of the Disappearing Barn Owl
Penn Vet’s Wildlife Futures Program and the Pennsylvania Game Commission (PGC) have been engaged in a collaborative effort to identify the causes of these owls’ decline and any actions that…

Penn Vet’s Annual Research Retreat Calls for the Power of Synergy in Challenging Times
Noting the challenges ahead, but celebrating the many breakthroughs at hand, the University of Pennsylvania School of Veterinary Medicine (Penn Vet) community gathered for their 31st annual Research Retreat held…


Penn Today News

Dogs with cancer are helping save lives—both canine and human (link is external)
The Comparative Immunotherapy Program led by Penn Vet’s Nicola Mason is redefining how therapies are developed and tested—uniting human and veterinary medicine to move promising immunotherapies forward.

Applying design to agriculture for a new approach to agroforestry (link is external)
Agriculture is a key sector of the U.S. economy, generating over $1 trillion annually and supporting millions of jobs. It’s also one of the country’s largest land uses; according to…

Keeping food safe and animals healthy (link is external)
A strain of the H5N1 virus—best known for causing avian influenza—was detected in U.S. dairy cattle for the first time in March 2024. It has since spread to more than…
In the Media




Bellwether Features

Penn Vet Holds Ribbon Cutting for New $2.8 Million Richard Lichter Advanced Dentistry and Oral Surgery Suite
The University of Pennsylvania School of Veterinary Medicine (Penn Vet) commemorated the official re-opening of the newly named Richard Lichter Advanced Dentistry and Oral Surgery Suite at Ryan Hospital with…
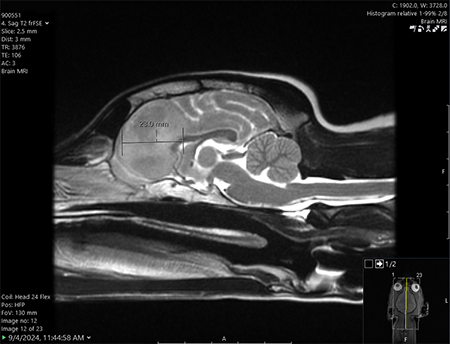
One Tiny Dog’s Outsized Contribution to Brain Surgery
Geddy Lee has lived a big life for a little dog. As a puppy, the tiny terrier mix was abandoned in Mississippi during a high-speed car chase. Rescued by law…

From Bird Fractures to Human Shoulders
As a child in Central Pennsylvania, Stephen Peoples, V’84, loved science and the natural world.

In the Office with Igor Brodsky, PhD
In his Philadelphia-based lab, Igor Brodsky is on a mission to understand how the body recognizes and responds to invading organisms that cause several diseases.
For the media
Press Inquiry
For all press inquiries, please the form below. This form is for media purposes only. If you are an animal owner, contact Ryan Veterinary Hospital for Companion Animals or, for our large animal hospital, New Bolton Center.
Non-News, Commerce-Affiliated Content Site Inquiries
The Office of Communications is selective in responding to requests from freelance writers on assignment for non-news, content building sites.
- All interviews with clinicians, faculty, staff and/or students in association with Penn Vet, Ryan Hospital, or New Bolton Center must be arranged through and facilitated by a representative from the Office of Communications. Please do not contact such individuals directly.
- When submitting a media or interview inquiry, please fill out this form. Include your name, contact information, and general details of your pitch including important deadlines.
- Visits to Penn Vet’s campuses and/or hospitals by members of the media must be arranged directly through a representative from the Office of Communications. For the well-being and privacy of our animal patients and their owners, a representative from the Office of Communications must also accompany any arranged visits.
- If an interview requires a Penn Vet clinician, faculty or staff member, or a student to be recorded on video, please be aware that a signed copy of the University’s video authorization form must be submitted to and confirmed by a representative from the Office of Communications prior to any taping. Additionally, an an Appearance Release must be supplied to the Office of Communications for review and approval prior to obtaining a signed agreement from the individual that is to appear on camera.